Research
, Volume: 19( 9)Did the Anthocyanin Colour System had Ancestors? What Physical Chemistry can Tell us
- *Correspondence:
- Pina F Department of Chemistry, Universidade Nova de Lisboa, Caparica, E-mail: fp@fct.unl.pt
Received: September 01, 2021; Accepted: September 15, 2021; Published: October 22, 2021
Abstract
Anthocyanins are the basis of the angiosperms colour, 3-deoxyanthocyanins and sphagnorubin play the same role in mosses and ferns and auronidins are responsible for the colour in liverworts. The separation of the ancestors of each of these lineages occurred in different periods of land plant evolution, therefore, our thesis is that chemical evolution of the colour systems accompanied plant evolution. The colour system of cyanidin-3-O-glucoside (kuromanin) as a representative compound of simpler anthocyanins was here fully characterized by stopped flow. This type of anthocyanins cannot confer significant colour to plants without intra or intermolecular interactions, complexation with metals or supramolecular structures as in Commelina communis. The anthocyanin’s colour system was compared with the one of 3-deoxyanthocyanins and riccionidin A the aglycone of auronidins. The three systems follow the same sequence of chemical reactions, but the respective thermodynamic and kinetics is dramatically different.
Keywords
Anthocyanins; 3-Deoxyanthocyanins; Auronidins; Plant evolution
Introduction
The thesis that we are presenting in this work is that the anthocyanin system that confers colour to angiosperms, starred by different molecules, was previously used by liverworts, mosses, and ferns. We mean by anthocyanin system, the flavylium cation, quinoidal base, hemiketal and, cis- and trans-chalcones, as well as the chemical reactions that interconvert them. The estimates for the origin of mosses fall within the Cambrian to Ordovician 1 and those of liverworts within the Ordovician to Early Devonian)2,3 but several competing hypotheses have hindered a consensual age estimate for these groups4. Ferns originated in the Early-Mid Devonian5 and angiosperms during the Lower Cretaceous, possibly earlier. The colour systems that have been used by these plants are based on furanoflavylium cations (liverworts), 3-deoxyanthocyanidins and sphagnorubin (mosses and ferns) and anthocyanins (angiosperms). The colour system of anthocyanins is unique in versatility when compared with the other two [1].
Colour is ubiquitous in Nature and anthocyanins are the largest class of water-soluble compounds present in the plant kingdom responsible for a range of colours in plants, from orange-red to purple-blue hues. Anthocyanins are glycosylated forms of anthocyanidins, normally occurring as 3-glycosides or 3,5-diglycosides, FIG.1. Different types of monosaccharides (e.g., glucose, galactose, rhamnose, and arabinose) and disaccharides (e.g., rutinose, sambubiose, and sophorose) can be attached to the flavylium core, and these sugars can also be acylated with cinnamic (e.g., caffeic, coumaric, ferulic, and sinapic acids) or aliphatic acids (e.g., acetic, malic, oxalic, and succinic acids).

Figure 1: The flavylium cation of Auronidins, 3-deoxyanthocyanins, sphagnorubin and anthocyanins.
Related with anthocyanins are 3-deoxyanthocyanidins, found in mosses and ferns, sphagnorubins found in peat moss, as well as auronidins (furanoflavylium derivatives), Andersen and Davies, questioned that “auronidins might contribute to the remarkable ability of liverworts to survive in extreme environments on land, and their discovery calls into question the possible pigment status of the first land plants.”
In the last two decades we have been devoted to the study of the kinetics and thermodynamics of the natural anthocyanins and 3-deoxyanthocyanins as well as related synthetic flavylium compounds. Following these studies, we investigate the kinetics and thermodynamics of furanoflavylium compounds and more recently riccionidin A, the aglycone of the auronidin reported by Andersen and Davies. In this work we argue about the three colour systems used by plants: auronidins, 3-deoxyanthocyanins and anthocyanins, giving some physico-chemical arguments to consider that the three colour systems in these families of plants accompanied their evolution. Some similarities and differences between the three systems were already reported by some of us. In this work, we focus essentially on the higher potentialities of the anthocyanin colour system when compared to the other two. The newly reported theoretical corpus based on the reverse pH jumps followed by stopped-flow that was used to characterize malvidin-3-O-glocoside (oenin) was applied to cyanidin-3-O-glucoside (kuromanin).
Results and Discussion
The colour system of anthocyanins. The case of cyanidin-3-O-glucoside (Kuromanin). The colour system of kuromanin is shown in FIG. 2.

Figure 2: Multistate of chemical species of the anthocyanin kuromanin in acidic medium. At sufficiently high proton concentrations the system converges to the flavylium cation. Kn=kn/k-n (n=a, h, t, i).
Flavylium cation is the sole species at pH≤1. When the pH is raised up to moderately acidic solutions (here on defined by direct pH jumps) other species are formed in the following sequence: quinoidal base, hemiketal, cis-chalcone and transchalcone. At higher pH values the anionic, or multi-anionic, analogues of the neutral species are formed, depending on the pH.
The kinetic processes towards the equilibrium after a direct pH jump is very illustrative. Proton transfer is by far the faster step of the kinetics and occurs during the mixing time of the stopped flow (few milliseconds). During the following steps AH+ and A behave as a single species with the respective ratio equal to [A]/[AH+]=Ka/[H+]. The next step is the formation of the hemiketal, B, through the hydration of AH+ (min), followed by the ring opening to give cis-chalcone, Cc, (ms/s). The fact that the quinoidal base does not open in acidic medium is a breakthrough discovery15 crucial for the comprehension of anthocyanins and related compounds. The isomerization to give trans-chalcone, Ct, in anthocyanins takes place in hours. When the system is equilibrated in moderately acidic pH values, addition of acid to reach pH ≤ 1 (reverse pH jumps) restores the flavylium cation. This is the basis for applying the reverse pH jumps followed by stopped flow [2].
The thermodynamic behaviour of anthocyanins can be represented by an energy level diagram, or by the mole fraction distribution as a function of pH. Both representations can be extended to the anionic forms obtained through deprotonation of the hydroxyl substituents in neutral and basic solutions. However, the energy level diagram becomes more complex and difficult of interpretation when the anionic species are included and by this reason is usually used to describe the equilibrium of the first row of Scheme 3.
Anthocyanins multistate.
The equilibrium involving anthocyanins and presented.
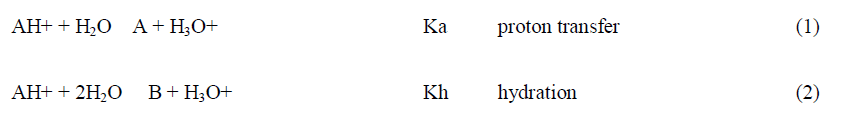
Extending to higher pH values

This complex system can be dramatically simplified to a polyprotic acid given by eq.9 and eq.12.
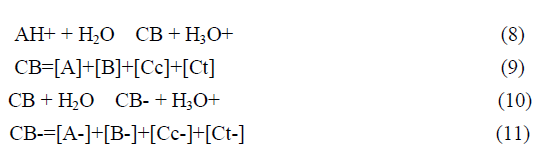
The fact that this complex system can be simplified considering the flavylium cation in equilibrium with its conjugated forms CB and CB- allows for the simplification of the mathematical treatment.
In anthocyanins and many other flavylium derived multistate of species the isomerization is by far the slowest process of the kinetics and a pseudo-equilibrium can be defined as a transient state obtained before significant formation of trans-chalcone.
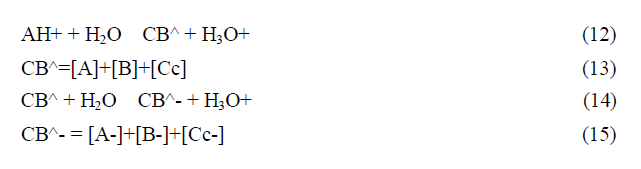
The most accurate method to achieve the equilibrium constants reported in Scheme, is based on the reverse pH jumps (defined by the addition of acid back to pH≤1 of equilibrated solutions, or pseudo-equilibrated, at higher pH values) 20 monitored by stopped flow.21 In the trace of a typical reverse pH jumps is shown. Three amplitudes are observed. The first is due to the flavylium cation absorption resulting from the conversion of all quinoidal bases (neutral and anionic) during the mixing time of the stopped flow together with some flavylium that is in equilibrium prior to the pH jump (for lower pH values). The amplitude of the faster kinetic step results from the conversion of hemiketal into flavylium cation, because at pH ≤ 1 the change of regime causes the hydration/dehydration to be faster than the tautomerization. Finally the last amplitude corresponds to the formation of more flavylium cation from cis-chalcone via hemiketal. Normalization of these amplitudes to the unit gives directly the mole fraction distribution of these species at the pseudo-equilibrium. The experimental data obtained for kuromanin in was used to calculate the equilibrium constants reported. Once obtained these constants, the energy level diagram can be constructed considering that, the Gibbs free energy, R perfect gases constant, T absolute temperature (Kelvin degrees) and K the equilibrium constant of the chemical reaction [3].
The mole fraction of Ct and Ct- is calculated by following the reverse pH jump through a standard spectrophotometer. Extending to a series of pH jumps from different pH values, the mole fraction distribution of Ct is calculated, and the mole fractions of AH+, A+A-, B+B- and Cc+Cc- are recalculated for the equilibrium discounting the one of Ct and Ct-. In Table 1 the equilibrium constants of kuromanin as well as the mole fraction distribution of all species at the equilibrium are shown. It should be emphasized that the constants regarding the anionic species have higher uncertainty because of the propagation of the errors law (depending on the equilibrium constants of the neutral species) and due to the instability of the simpler anthocyanins at higher pH values, in particular to determine the mole fractions of Ct.
Limits of anthocyanins to confer colour
Anthocyanins are located in the vacuoles. The vacuoles pH may vary considerably, but a pH of approximately 5 is often observed.24 However, the pH of the vacuoles could be as low as 2.0 in citrus fruits25 and 7.7 or more in some flowers.26 In anthocyanins the coloured species are flavylium cation and quinoidal bases. In the case of simple anthocyanins like malvidin-3-O-glucoside (Oenin) and cyanidin-3-O-glucoside (kuromanin) the appearance of colour at the equilibrium is only observed for the flavylium cation at very low pH values and at moderately acidic pHs the mole fraction of the quinoidal base is low. Consequently, neither flavylium cation nor quinoidal base can confer significant colour at moderately acidic solutions. Curiously, at pH values close to neutrality the colour of the anionic quinoidal base is accessible but at these pH values these anthocyanins are unstable. It is also interesting to note that, despite the generally accepted view by the scientific community and contrary to what can be observed for oenin, the ionized quinoidal base (A-) of kuromanin seems to not confer the blue colour to the solution.
To investigate the phenomenon in more detail, a series direct pH jumps from pH = 1 to higher pH’s values (up to pH =12) were performed by stopped-flow to calculate the four deprotonation reactions. This technique allows for the fast (ca. 10 ms after the pH jump) acquisition of the absorption spectra which are necessary to investigate the acid-base equilibria without interference of the hydration and subsequent reactions that leads to colour fading. The plots of absorbance versus pH, at four different wavelengths, that were used to obtain the respective pKa values for the formation of the quinoidal base and its ionized forms from the flavylium cation.
Using this methodology the following values were obtained: pKa1=4.0; pKa2=6.5; pKa3=10.0 ; pKa4=12.2 which compares with the ones obtained for oenin: pKa1=3.9; pKa2=6.3; pKa3=8.4. While the first two are reasonably similar, the pKa3 for the formation of the di-ionized quinoidal base is considerable higher for kuromanin. By combining the pH-dependent mole fraction distribution of the flavylium cation and all quinoidal bases with the respective spectral data, it is possible to obtain the spectra of the five species by spectral decomposition.
The results clearly show that the spectrum of the A- cannot account for the blue color (in contrast with A- of oenin which is red-shifted) and that this color can only be expressed by A2- and A3- which appear only at very basic pH values. Since these conditions does not seem feasible in biological systems, the results suggest that the kuromanin can only express the blue color when complex with metal cations or by other mechanism that may shift the third and fourth pKa to lower values.
In order to overcome the limits of simpler anthocyanins to confer colour, Nature used different strategies. One of the most studied is co-pigmentation, defined by Robinson as the modifications of the anthocyanins absorption spectrum caused by colourless compounds, such as amino acids, sugars and flavonoids. Co-pigmentation could be intermolecular and/or intramolecular as observed in acylated anthocyanins. Other strategies used by Nature are self-association and metal complexation. Summarizing, the stabilization of the coloured species is usually driven by several noncovalent interactions, such as hydrogen bonding, van-der-Waals, metal-ligand interactions and hydrophobic driven associations.
The magnitude of thee rates of the inter-conversion between the several species of the anthocyanins multistate. After a direct pH jump for example to pH=6, quinoidal base is formed, with a rate that is faster than the mixing time of the stopped-flow, requiring other techniques, such as temperature jumps15 or flash photolysis35 to be observed. In the consecutive kinetic steps AH+ and A are in equilibrium, the respective ratio equal to [A]/[AH+]=Ka/[H+]. The disappearance of AH+ and A, towards the equilibrium is a bi-exponential process, 2nd and 3rd steps, exhibiting very different lifetimes, minutes and hours respectively. Since at the pH values reached in direct pH jumps the tautomerization is faster than the hydration the second step is controlled by the last.
The expression that accounts for the 2nd step was achieved in a straightforward manner considering that the species AH+ and A on one side and B and Cc on the other are in fast equilibrium during the hydration step.
Here, and are the mole fraction of AH+ in its equilibrium with A and the mole fraction of B in its equilibrium with Cc, respectively. At the end of the 2nd step the system reaches the pseudo-equilibrium. The 3rd step is controlled by the isomerization and due to the difference between the rates between of the 2nd and 3rd steps, AH+, A, B and Cc can be considered in equilibrium.
The mole fraction of Cc at pseudo-equilibrium. Some conclusions may be taken regarding the thermodynamics and kinetics of the anthocyanins system. At the equilibrium, the red, pink and blue colours are potentially available, but some strategies should be used by the plants to give expression to these colours; intramolecular and/or intermolecular co-pigmentation in acylated anthocyanins and supramolecular structures like the one that gives the blue colour to Commelina communis, as reported by Nagoya’s group. The colour system of 3-deoxyanthocyanins 3-deoxyanthocyanins follow the same multistate of species of anthocyanins, FIG. 3, but the isomerization is not the slowest kinetic step towards equilibrium [4].

Figure 3: Kinetic scheme of luteolinidin.
In a direct pH jump AH+/A conversion into Ct occurs via a single step of pseudo-first order kinetics. Considering AH+ in fast equilibrium with A (X) on one side and B in fast equilibrium with Cc (Y) on the other to produce Ct (Z), a mechanism equivalent to a reversible kinetic scheme involving 3 species is required. When the steady state approach is applied to Y. For luteolinidin and related compounds, the rate constants of the reverse pH jumps are equal to the direct ones for the same final pH. Moreover, B and Cc are elusive species appearing at unmeasurable concentrations in the stationary state. However, the mole fractions of A and Ct can be calculated from the data of the bell-shaped curve to obtain the respective mole fraction distribution from the ratios Ka/K’a and KhKtKi/K’a.
The comparison between response to the kinetics of the colour appearance and disappearance is discussed in the next section. No blue colour can be achieved by 3-deoxyanthocyanins.Comparison of the colour system in anthocyanins and 3-deoxyanthocyanidins. The chemical structure of the cyanidin, the aglycone of kuromanin (anthocyanidin), when compared with luteolinidin shows that they differ only in the hydroxyl in position 3. It is worth of note that cyanidin, like the other anthocyanidins, is very unstable at moderately acidic solutions,39 while luteolinidin40 and sphagnorubin A9, both found in mosses, are relatively stable. This suggests that it was not because of the stability that the hydroxyl was introduced in position 3 of anthocyanins. The reason was that this substitution gives rise to a red flavylium cation, a pink quinoidal base and a bluish or blue (depending on the anthocyanin) anionic quinoidal base. The unstability of anthocyanidins was solved by glycosylation of position 3 to give 3-O-anthocyanidins (anthocyanins). Glycosylation of position 5 that gives the diglucosilated anthocyanidins is not so significant for the stability.
The quinoidal base of luteolinidin is red and the anionic quinoidal base is orange. There is no blue colour. The blue colour seems to be the most demanding in terms of the evolution. The excellent review by Yoshida et al. gives an overview of the structures used to express this colour.38 Intermolecular and intramolecular co-pigmentation (in acylated anthocyanins) and metal complexation are the strategies used to get blue colour. The most sophisticated structures (from the chemical point of view) are metaloanthocyanins, as for example the one that confers colour to Commelina communis (FIG. 4). In this supramolecule, 2 metals, 6 anthocyanins and 6 flavanones are organized in two parallel plans [5].

Figure 4: Sketch of the metalloanthocyanin responsible for the colour of Commelina communis.
The sketch was provided by Prof. Kumi Yoshida. The building blocks self-associate to produce the supramolecule in a bottom-up approach. Another difference in both systems is the pH dependent mole fraction distribution of the chemical species appearing at the equilibrium and the respective kinetics. When the plants need to increase or decrease the colour to protect from the environment, anthocyanins have a reservoir of the colourless hemiketal (in fast equilibrium with cis-chalcone) that quickly increase or decrease the amount of colour through the hydration/dehydration reaction. In contrast, 3-deoxyanthocyanins have an equilibrium between the coloured species flavylium cation and quinoidal base and trans-chalcone. The appearance/disappearance of colour takes place through the interconversion AH+/A and Ct and follows the kinetic curve, while in anthocyanins is the much faster hydration reaction that controls.
Auronidin-2’-neohesperidoside was extracted from the liverwort Marchantia polymorpha, by Davies and Andersen10 who coined these molecules as auronidins. In recent years we studied the thermodynamic and kinetics of the so-called furanoflavylium compounds, a term used by Chakravarty to designate the compounds (FIG.5)

Figure 5: 4’-hydroxyfuranoflavylium,11 4’,7-dihydroxyflavylium,12 and riccionidin A, the aglycone of auronidin-2’-neohesperidoside,13 follow the same multistate of species of anthocyanins and 3-deoxyanthocyanins.
We verified that in 4’-hydroxyfuranoflavylium, 4’,7-dihydroxyfuranoflavylium, and riccionidin A, the aglycone of auronidin-2’-neohesperidoside, follow the same multistate of species of anthocyanins and 3-deoxyanthocyanins. Riccionidin A was identified in several liverworts as for example in the Antarctic Cephaloziella varians, in response to an abrupt increase of UV-B radiation,42 in in vitro cultures of Ricciocarpos natans, as well as in adventitious root cultures of the Anacardiaceae Rhus javanica (FIG. 6).

Figure 6: Riccionidin follows the same multistate of anthocyanins and 3-deoxyanthocyanins.
The most interesting feature of riccionidin A is its extremely slow rate to convert trans-chalcone in flavylium cation. In acetic acid saturated with HCl gas at 100ºC the conversion requires more than 11 days, At pH=5.0 in methanol:water (1:1) at 45ºC, the reaction is again very slow, but the acidity is not enough to give flavylium cation.
It was possible to identify the species AH+, A, B, Cc and Ct by HPLC/MS13 before decomposition, confirming that the multistate of species is the same of anthocyanins and 3-deoxyanthocyanins. Moreover, the degradation products of these three families seem to be similar.
The degradation process of anthocyanidins starts with the keto-enol equilibrium involving C3-OH in hemiketal and chalcones species. The formation of the species from the chalcones allows the easily oxidative C-ring fission of the molecule by the cleavage of the C2-C3 or C3-C4 bonds yielding two different degradation products Glycosylation of C3-OH (anthocyanins) prevents this keto-enolic equilibrium and enhance the stability of the molecule. For anthocyanins, the degradation process starts with the breaking of the glycosyl bond releasing the OH group at C3. The system behaves from that point as an anthocyanidin. Moreover, the absence of OH group at position 3 (3-desoxyanthocyanidins) also enhance the stability of the molecule, although some degradation product could be detected as minor component. In that sense, the transchalcone formed is very stable and its degradation by oxidative dimerization or by C-ring fission is much less extended than in anthocyanidins.
Auronidins also shows a similar degradation behavior. In this case, the trans-chalcone is less stable than before and can be degraded by oxidative coupling, being this species the major degradation product detected by HPLC/MS. Moreover, the furan ring could also perform a ring opening that allows the C-ring fission degradation as minor products. The data shown above and previously reported by some of us 13 is a proof that riccionidin A follows exactly the same multistate of species and chemical reactions of anthocyanins and 3-deoxyanthocyanins.The colours that are available in riccionidin. The photos were taken immediately after a direct pH jump. The blue colour is achieved only in extremely high pH values not found in plants [6].
As described above in riccionidin as well as in its glycoside form reported by Davies and Andersen, the flavylium cation is yellowish and the quinoidal base red. The stability of riccionidin A is limited to pH<4 and the rate constants between the species is extremely slow. At higher pH values, riccionidin. A starts its degradation following a similar pathway of anthocyanidins and 3-desoxyanthocyanidin. In that sense these three families of compounds yield the same products with higher or lesser extension depending on the relative stability of the chalcone species. All these three families are stable in acidic condition but when the pH increases, this is when the molar fraction of the chalcone increases, the degradation products start to appear in most cases by oxidative coupling or by C-ring fission degradation. As a suggestion for future work it would be interesting to verify if auronidins are able to form associations by co-pigmentation or give metal complexes, because there is a catechol in ring a could link to metals. The furano ring prevents the attachment of the sugar in position 3 and the glycosylation occurs in position. Anthocyanins like synthetic flavylium compounds bearing an hydroxyl in position 2’, are able to form flavanones from the mono-anionic chalcones. Another perspective for future studies would be to compare the Davies and Andersen auronidin, with the results obtained for riccionidin.
The direct pH jumps were carried out by mixing a stock solution of Kuromanin in HCl 0.1 M (3 × 10-5 M) with a solution containing NaOH 0.1M and Theorell and Stenhagen universal buffer at the desired final pH using the stopped flow (S × 20, Applied Photophysics; Surrey, UK) spectrometer equipped with a PDA/UV photodiode array detector. The direct pH jumps were also carried out adding a small aliquot of a concentrated stock solution of the anthocyanin (pH=1) into a 3 mL cuvette with water, enough NaOH to neutralize the stock solution’s acid, and universal buffer at the desired final pH. Spectroscopic measurements were performed using Mili-Q water at a constant temperature of 20 ± 1ºC using a Varian-Cary 100 Bio or Varian. Cary 5000 spectrophotometers. Reverse pH jumps were carried out, by stopped flow (pseudo-equilibrated solutions) and common spectrophotometer, adding enough HCl to reach pH=1 in equilibrated solution of the anthocyanidin/anthocyanin at different pH values. The final pH of the solutions was measured in a Radiometer Copenhagen PHM240 pH/ion meter.
Conclusion
The physical chemistry of riccionidin A and 3-deoxyanthocyanins shows unequivocally that the former is much less versatile than the last. Anthocyanins while limited a priori to express the colour by themselves have a complete colour pallet available and can give expression to these colours by means of intra and intermolecular co-pigmentation, coordination to metals and all these effects combined as in the case of metaloanthocyanins. There is no doubt that the colour systems had also an evolution from auronidins, 3-deoxyanthocyanins and anthocyanins and that this evolution seems to be parallel to the evolution of the respective plants. We wonder if the results reported in this work could contribute to the current discussion of the phylogenetic hypotheses on divergence between mosses and liverworts, and between this group and vascular plants.
References
- Biolley JP, Jay M. Anthocyanins in modern roses: Chemical and colorimetric features in relation to the colour range. J Experim Bot. 1993;44(11):1725-1734.
- Pina F, Melo MJ, Laia CA, et al. Chemistry and applications of flavylium compounds: A handful of colours. Chem Soc Rev. 2012;41(2):869-908.
- Ogutcen E, Durand K, Wolowski M, et al. Chemical basis of floral color signals in gesneriaceae: the effect of alternative anthocyanin pathways. Front Plant Sci. 2020.
- Rudall PJ. Colourful cones: How did flower colour first evolve?. J Experim Bot. 2020;71(3):759-767.
- Saona CR, Cloonan KR, Pedraza FS, et al. Differential susceptibility of wild and cultivated blueberries to an invasive frugivorous pest. J Chem Ecol. 2019;45(3):286-297.
- Streisfeld MA, Rausher MD. Altered trans-regulatory control of gene expression in multiple anthocyanin genes contributes to adaptive flower color evolution in Mimulus aurantiacus. Molecul Biol Evol. 2009;26(2):433-444.
